|
|
|

|
Start by activating the ‘Show mass values in graph’ button (fifth in the toolbar). This opens the ‘Mass vs. precision’ graph. |
||||||||||||
 |
 |
|||||||||||
|
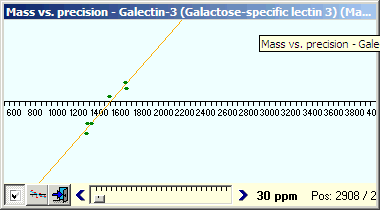
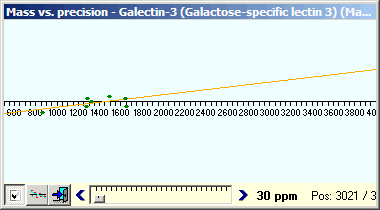
The graph (left above) shows all the ‘marked’ peptides in the peptide hit list (In the peptide ‘hit’ list, all perfect matches are initially checked in the left-hand column while other ‘potential hits’ are unmarked). You may already have found peptides that were not detected in your initial PMF search, as these searches are usually performed using a missed cleavage value of ‘1’ and GPMAW displays all peptides fitting the enzyme cleavage definition (using a higher value in PMF will not increase score as the relative database will increase more than the few extra hits increase the score). To increase the number of hits and coverage click on the E button in the peptide hit toolbar to change it to a C (check fit). This removes the constraint that peptide hits have to fit the exact enzyme cleavage specifications and will thus show all peptides that fits within the given mass window. |
||||||||||||
 |
||||||||||||
|
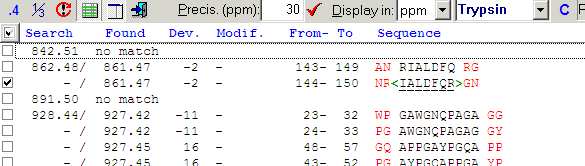
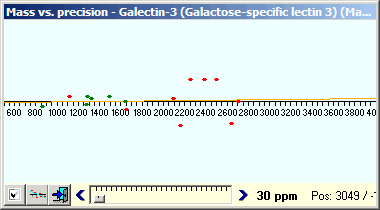
As can be seen, the number of potential hits is large and difficult to interpret. Change the search precision to 10 ppm (remember the ‘known’ hits are within 2 ppm) to limit the choices. Change focus back to the graph, unpress the left-hand button to see all peptide hits (exact fits are green, others are red, modification yellow - none here). |
||||||||||||
 |
||||||||||||
|
Four of the red dots are within the same limits as the green ones and five are outlyers and can be discarded. Looking at the sequences of these hits, the first one shows tryptic C-terminal cleavage and N-terminal cleavage after Tyr. As this is a known secondary cleavage site for trypsin it means that it may be acceptable. Of the other three peptides one shares the mass with a ‘perfect’ hit, one show cleavage after Gly and the last has no tryptic cleavage at all and all three are thus discarded. The results of the analysis can now be viewed in the report - click on the ‘Report’ tab at the bottom of the window to show the Search report. |
||||||||||||
|
Site last updated: February 14, 2025 |
||||||||||||