|
|
|

|
Teh search report is a dual window with the protein sequence at the top and description of the search below. The ratio of the two windows can be changed by grabbing the green divided with the mouse and slide it up and down. |
|||||||
 |
|||||||
|
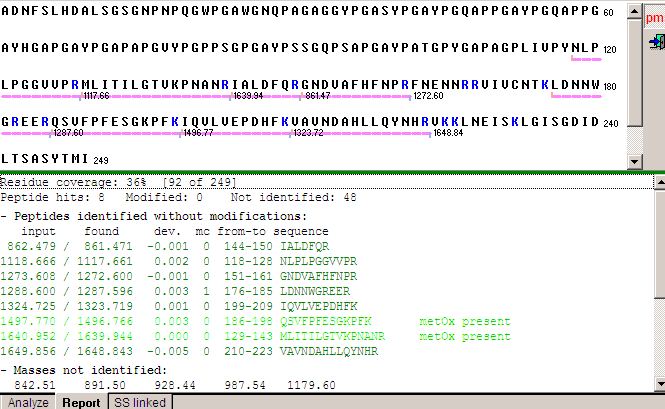
Peptides found in the search are underlined, and the mass of each peptide shown to the right of each underline. Only peptides in the C-terminal part has been identified, and the ‘semi-tryptic’ peptide we found fits snugly as the first peptide in the series, further corrobation of the identification - actually, as no basic residues are present in the first half of the sequence we would not expect to see any peptides from this part, effectively doubling the residue coverage reported (i.e. observed peptides / possible peptides). Furthermore, the remaining peptides in the C-terminal region are small (hard to detect) except for the C-terminal peptide which was not observed. In the case of a C-terminal trimming, the hit list on the previous page could be searched for peptides starting with LGIS - but none were found. As this was a tryptic digest, the pmf button in the top right corner was depressed resulting in coloring of the found peptides according to the following rules:
You may switch back and forth between the Analyze and the Report pages to check and verify and make more adjustments to your search. As you analyze more and more mass spectra you will find that GPMAW will help you to extract just that little bit of extra information. |
|||||||
|
Site last updated: February 14, 2025 |
|||||||